
Technology Overview
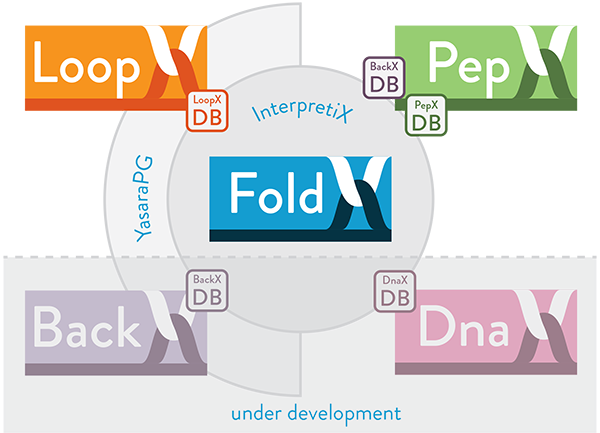
The FoldX Suite
The FoldX Suite builds on the strong fundament of advanced protein design features already implemented in the oldest FoldX versions and integrates new capabilities: loop reconstruction (LoopX) and peptide docking (PepX). The Suite also features an improved usability thanks to a new boost Command Line Interface.
The FoldX Suite exploits the power of fragment libraries [bib]215[/bib], by integrating in silico digested backbone protein fragments of different lengths. Such fragment-based strategy is implemented by means of two databases: (i) BackXDB, a database containing fragments from over 7000 non-homologous proteins from the Astral collection, segmented in lengths from 4 to 14 residues and clustered according to structural similarity, summing up to a content of 2 million fragments per fragment length; (ii) LoopXDB, a database of non-regular structure elements, clustered according to end-to-end distance between the regular residues flanking the loop. This structural information is used in order to mimic backbone moves and model loop flexibility on crystal structures.
The Suite Main Core - FoldX
FoldX provides a fast and quantitative estimation of the importance of the interactions contributing to the stability of proteins and protein complexes. The predictive power of FoldX has been tested on a very large set of point mutants (1088 mutants) spanning most of the structural environments found in proteins. FoldX uses a full atomic description of the structure of the proteins. The different energy terms taken into account in FoldX have been weighted using empirical data obtained from protein engineering experiments. The present energy function uses a minimum of computational resources and can therefore easily be used in protein design algorithms, and in the field of protein structure and folding pathways prediction where one requires a fast and accurate energy function.
The Force Field
The FoldX energy function includes terms that have been found to be important for protein stability. The free energy of unfolding (ΔG) of a target protein is calculated using the equation:
ΔG=Wvdw⋅ΔGvdw+WsolvH⋅ΔGsolvH+WsolvP⋅ΔGsolvP+ΔGwb+ΔGhbond+ΔGel+ΔGKon+Wmc⋅T⋅ΔSmc+Wsc⋅T⋅ΔSsc
where ∆Gvdw is the sum of the van der Waals contributions of all atoms with respect to the same interactions with the solvent. ∆GsolvH and ∆GsolvP are the differences in solvation energy for apolar and polar groups respectively when these change from the unfolded to the folded state. ∆Ghbond is the free energy difference between the formation of an intra-molecular hydrogen bond compared to inter-molecular hydrogen-bond formation (with solvent). ∆Gwb is the extra stabilising free energy provided by a water molecule making more than one hydrogen bond to the protein (water bridges) that cannot be taken into account with non-explicit solvent approximations[bib]189[/bib]. ∆Gel is the electrostatic contribution of charged groups, including the helix dipole. ∆Smc is the entropy cost of fixing the backbone in the folded state; this term is dependent on the intrinsic tendency of a particular amino acid to adopt certain dihedral angles[bib]190[/bib]. Finally ∆Ssc is the entropic cost of fixing a side chain in a particular conformation[bib]191[/bib]. While working with oligomeric proteins or protein complexes, two extra terms are involved: ∆Gkon which reflects the effect of electrostatic interactions on the association constant kon (this applies only to the subunit binding energies)[bib]193[/bib] and ∆Str which is the loss of translational and rotational entropy that ensues on formation of the complex. The latter term cancels out when we are looking at the effect of point mutations on complexes. The energy values of ∆Gvdw, ∆GsolvH, ∆GsolvP and ∆Ghbond attributed to each atom type have been derived from a set of experimental data, and ∆Smc and ∆Smc have been taken from theoretical estimates. The terms Wvdw, WsolvH, WsolvP, Wmc and Wsc correspond to the weighting factors applied to the raw energy terms. They are all 1, except for the van der Waals’ contribution which is 0.33 (the van der Waals’ contributions are derived from vapor to water energy transfer, while in the protein we are going from solvent to protein). For a detailed explanation of the FoldX force field, see[bib]197[/bib][bib]198[/bib] and the FoldX webserver[bib]196[/bib].
Mutating Proteins and DNA
FoldX can mutate the 20 natural aminocids, the phosphorylated versions of Ser, Thr and Tyr, the sulfated version of Try, methylated Lys, hydroxyl Proline and the standard 4 DNA bases, methylated adenosine and guanosine. When mutating DNA the code automatically identifies the pairbase of the selected base and mutates both of them simultaneously to keep the base pairing. In the case of DNA we do not use rotamers but we allow the base to move with respect to the ribose.
LoopX - Loop Reconstruction

LoopX allows fast and accurate prediction of protein loop structure by means of LoopXDB, a library of non-regular structure elements, clustered according to end-to-end distance between the regular residues flanking the loop. We have shown that proteins can be reconstructed by superposition of peptide fragments [bib]216[/bib]. Similarly, the LoopX algorithm allows to replace loops by other existing loops by in order to find variability while engineering proteins. Loop backbone templates are selected from the LoopXDB database that contains 14.525 protein structures with < 95% sequence identity. Anchor groups (2 residues, one loop and one non-loop residue) are chosen and fitted with all LoopXDB super classes by superposition on the super class centroids. To speed up this calculation, only loop super classes that have the same secondary structure residues embracing the loop are considered. A super class is accepted as donor of candidate loops when the RMSD of the backbone atoms (N, CA, C, O) between the four anchor residues of its centroid and the target loop after super- position is < 1.5 A.
PepX - Peptide Docking
PepX allows fast and accurate prediction of peptide docking by means of BackXDB, a library of protein building blocks, and PepXDB, a library of protein-peptide binding motifs
The PepX algorithm takes a peptide sequence and a domain structure as input and determines the accessible conformational space for bound peptides through interaction constraints with anchor fragments. A heuristic algorithm (CSP) is applied in order to reduce the conformational search space of all possible overlapping fragments of the PepXDB database. PepXDB is a database of peptide-peptide interactions that contains more than 7000000 interactions from 1431 PDBs, representing the diversity of structural information on peptide-peptide complexes available in the Protein Data Bank. For ab initio predictions we select a number of solutions as seeds from the most favorable fragment anchors. Intermediate solutions are further refined by local sampling with superimposed BackXDB fragments of the same length or longer lengths in the case of extension. Finally, the entire set of solutions is clustered by an unsupervised method that predicts the binding site and peptide conformation by analyzing cluster attributes.
BackX - Backbone Moves (Available soon in ModelX toolsuite)
Protein backbones are flexible and move allowing induced fit of the protein interfaces. Mimicking this flexibility can be achieved by means of small protein fragment superposition over anchor points, resulting in modelling backbone moves. The module is currently under development.
DnaX – DNA docking (Available in ModelX toolsuite)
The number of Protein-DNA crystal structures in a complex deposited in the PDB have increased exponentially during the last 25 years. This tool aims to predict DNA docking into protein by using the structural information stored in these crystal structures. The approach for DNA binding prediction we use is based on the assumption that Prot-Dna interactions occurs in a limited number of conformations. With this purpose in mind, we have digested in silico more than 2500 Prot-Dna crystal structures and built a repository of Prot-Dna interactions, classified by their structural information. This utility is available within the ModelX modeling toolsuite, please visit modelx.crg.eu.
InterpretiX
The Interpretix Smart Engine is a new command line interface that allows encapsulation of the different software packages of the FoldX Suite. It features:
- C++/Boost Command Line Interface
- Unified Command Line Interface and executable to launch different modeling tools
- Better error reporting of any problems with the command line
- Unified human-readable configuration file to work with different modeling tools (no more ‘XML-like’ syntax)
- High level programming layer, to combine different FoldX commands in a flexible and iterative way to generate complex analysis
- Interactive help on general and specific parameters
- Integration of a new FoldX Driver for parallel usage on cluster supporting the Java Sungrid Engine
YasaraPG
The FoldX Suite features a dedicated graphical interface to Yasara.
A Note on Naming
The FoldX Suite has required an intensive redesign of the structure of FoldX and its related packages and databases. Past users can refer to the following schema for naming conversion:
| Old Name | New Name |
|---|---|
| Brix, FragBrix | BackXDB |
| LoopBrix | LoopXDB |
| InteraX | PepXDB |
| BrixCluster | ClusterX |
| FoldX4 | FoldX3 + InterpretiX |